| MWexperimental | 14 | kDa |
| MWexpected | 14 | kDa |
| VPorod | 19 | nm3 |
|
log I(s)
1.05×100
1.05×10-1
1.05×10-2
1.05×10-3
|
 s, nm-1
s, nm-1
|
|
|
|

|
|

|
|

|
|

|
|

|
|
The consensus SAXS profile for lysozyme was generated by the datcombine tool (ATSAS 3.1.0) with both outlier- and error-filters applied. The data input to datcombine were ten scattering profiles made up of pure SEC-SAXS (1), pure batch SAXS (4) and merged SEC-SAXS-batch SAXS (5) data. All contributing data were independent measurements, and no individual measurement was represented more than once in the contributing scattering profile set. The buffer for substantial majority of the contributing data was 50 mM sodium citrate, pH 4.5, 150 mM NaCl. Protein concentrations for batch measurements ranged from 0.75 - 13.4 mg/mL, and all batch data that showed evidence of aggregation or interparticle interference were merged with SEC-SAXS or lower concentration data to remove any influence from aggregate. The starting lysozyme atomistic model for CRYSOL, Pepsi-SAXS, and FoXS calculations was the PDB ID 2VB1 with small-molecule crystallisation agents removed. Custom WAXSiS calculations (with Gromacs software) used the same coordinates and added explicit waters and appropriate number of ions for the MD calculations.
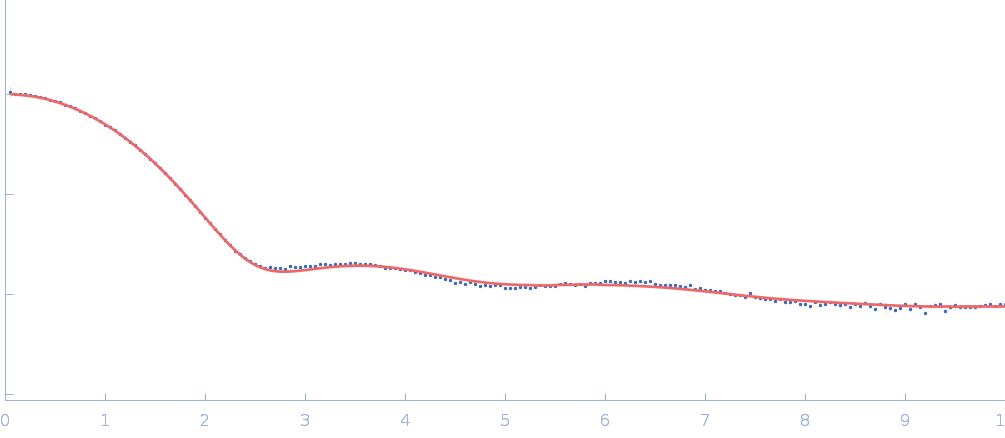
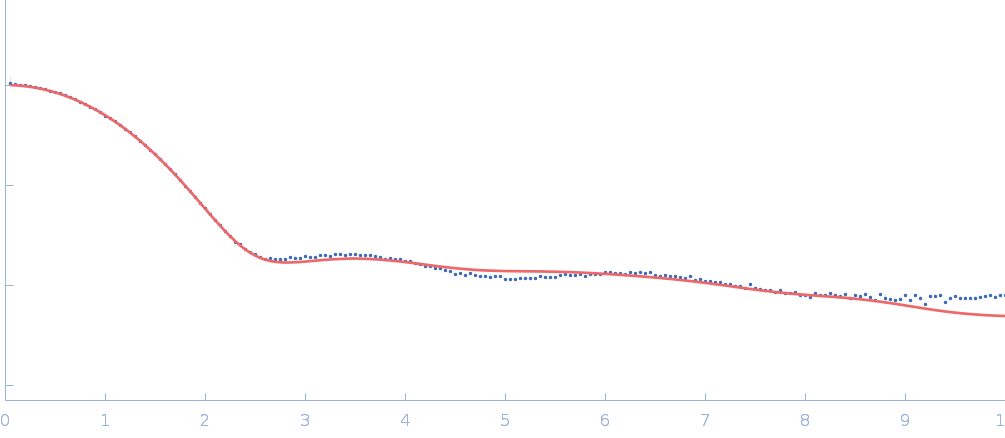
The data input to datcombine are made available for download in the associated zip file. Model fits are shown in order (top to bottom): DAMMIN, CRYSOL, Pepsi-SAXS, FoXS, and custom WAXSiS. The unusually good statistics for the consensus SAXS data generally give rise to large χ-square values for the model fits.
Tags:
benchmark
|
|
|||||||||||||||||||||||||||||||||||||||