|
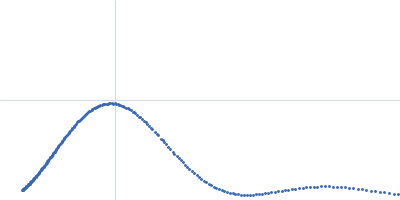
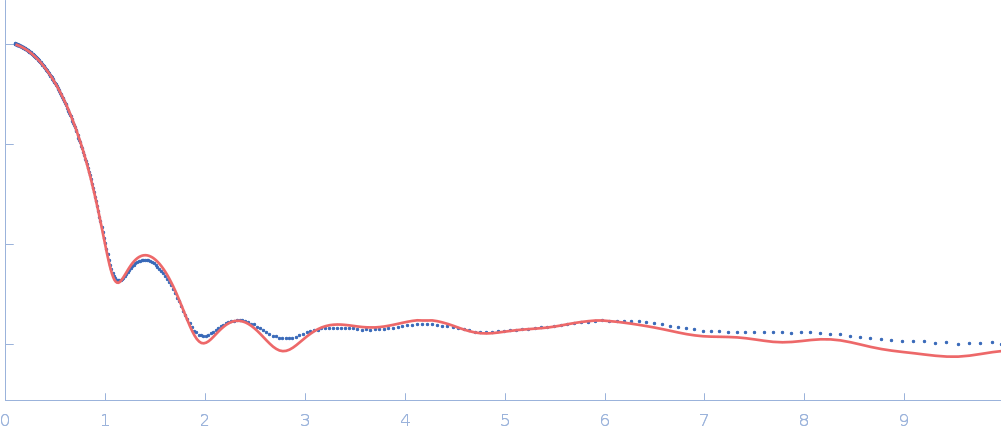
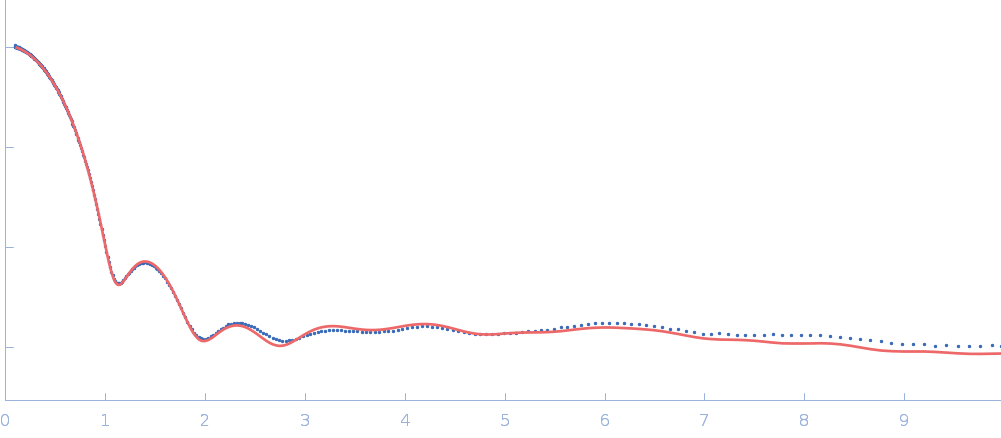
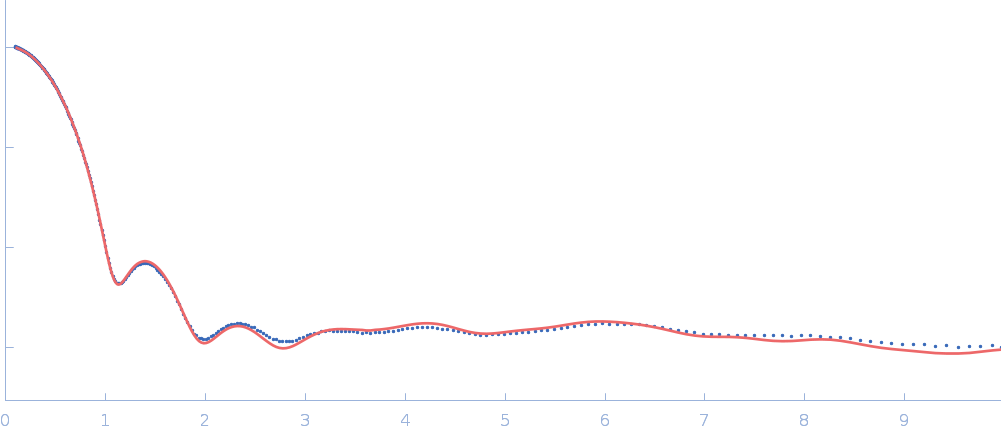
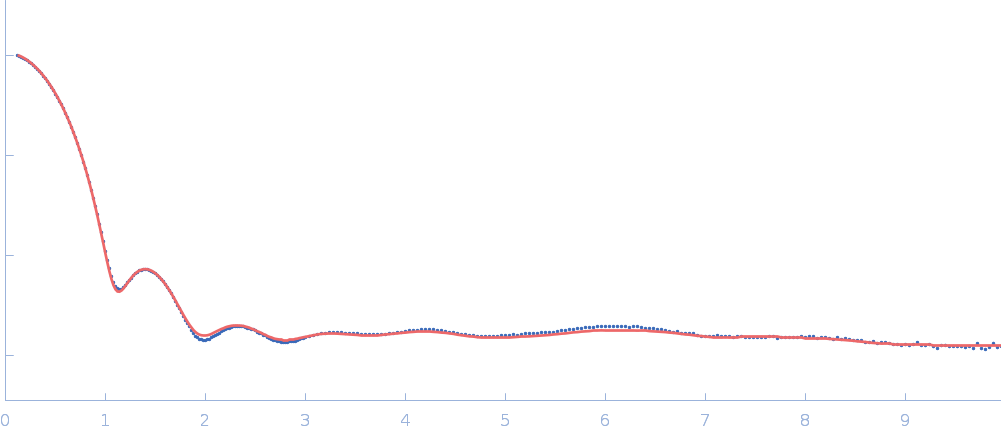
Updated consensus SAXS profiles generated using the ML-SAScombine tool (with log-s binning) for xylose isomerase in 50 mM Tris pH 7.5, 100 mM NaCl with 1 mM MgCl2. A total of 29 independent SAXS profiles (6 SEC-SAXS and 23 batch SAXS) contributed from 10 SAXS beamlines were combined. The minimum s value for SEC-SAXS data was 0.01 nm-1. Protein concentrations for batch measurements ranged from 1 - 20 mg/mL, and the minimum s value for these data (no less than 0.8 nm-1) was chosen to ensure exclusion of low-s data points affected by aggregation or interparticle interference. The xylose isomerase atomistic models for CRYSOL, Pepsi-SAXS, and FoXS calculations is the PDB ID 1MNZ with small-molecule crystallisation agents removed, and an N-terminal Met added using PyMol to reflect the sequence for the protein as measured by SAXS. Custom WAXSiS calculations used the same coordinates with added explicit waters and ions to match the experimental conditions for the MD simulations. WAXSiS calculations include statistical errors and the error weighting for residual differences is therefore the square root of the sum of the squares of the experimental and WAXSiS statistical errors. Model fits are shown in order (top to bottom): CRYSOL (classic directional hydration layer), FoXS, Pepsi-SAXS and custom WAXSiS. All three model fits with implicit hydration layer are to data on a log s-scale, while for custom WAXSiS the data are on a linear s-scale. The unusually good statistics for the consensus SAXS data generally give rise to large χ-square values for the model fits.
Additional data and information are made available in the full-entry zip archive and include: i) The input data for ML-SAScombine; ii) Runscripts used with ML-SAScombine; iii) Output files for updated consensus files from ML-SAScombine with log and linear s-binning; iv) Output files for combined SEC-SAS data from ML-SAScombine with log s-binning and; v) The original custom-WAXSiS model-fits with errors with the consensus data on the same s-grid.
|
|
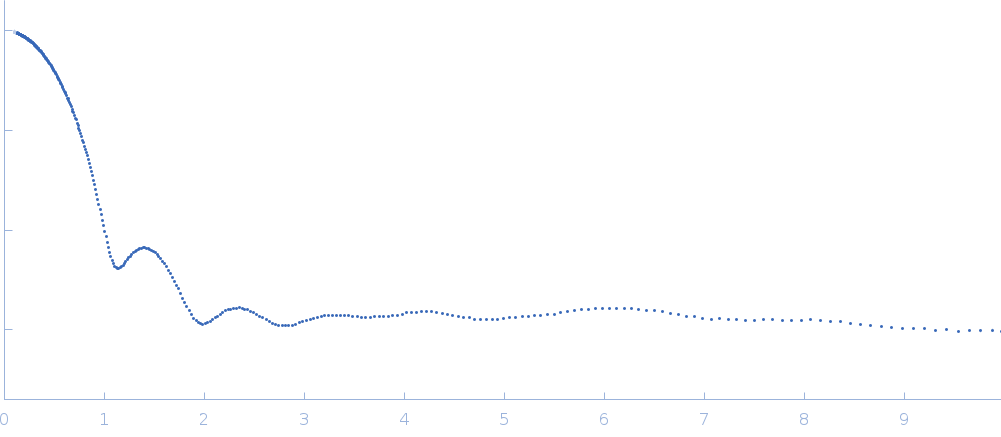 s, nm-1
s, nm-1