|
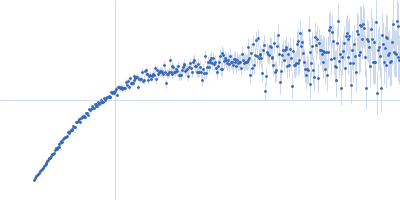
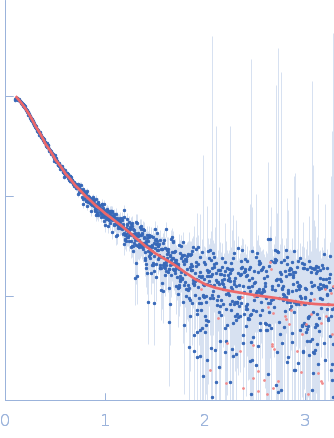
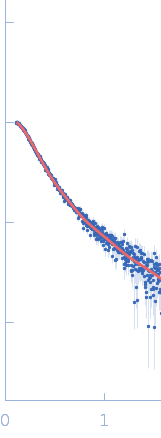
Synchrotron SAXS data from solutions of full-length myotilin fused to an N-terminal thioredoxin tag in 20 mM Tris, 400 mM NaCl, 250 mM arginine, 5% v/v glycerol, pH 7.5 were collected on the EMBL P12 beam line at PETRA III (Hamburg, Germany) using a Pilatus 2M detector at a sample-detector distance of 3.1 m and at a wavelength of λ = 0.124 nm (I(s) vs s, where s = 4πsinθ/λ, and 2θ is the scattering angle). In-line size-exclusion chromatography (SEC) SAS was employed. The SEC parameters were as follows: A 100.00 μl sample at 7.3 mg/ml was injected at a 0.50 ml/min flow rate onto a GE Superdex 200 Increase 10/300 column at 20°C. As described in Graewert et al. (Scientific Reports volume 5, Article number: 10734 (2015)), an integrated micro splitter (P-451 Upchurch Scientific) allowed the elutent of a chromatography column (Superdex 200 10/300 increase, GE) to flow equally through the SAXS capillary and a modular triple detector array (TDA, Viscotek model TDA 305, Malvern Instruments) that extracts MW estimates of SEC-separated components by correlating refractive index (RI) and/or UV-vis concentrations with right angle laser light data (RALS) using the integrated Omnisec software.In parallel. With this the expected MW of the monomer could be confirmed (70 kD). 3000 individual SAXS frames were collected with 1 s exposure that were used for subsequent SAXS analysis. SAXS data reduction to produce the final scattering profile of monomeric full-length Trx-Myot was performed using SASFLOW pipeline for radial averaging. CHROMIXS was used for the visualization and reduction of the SEC-SAXS dataset to produce the final subtracted curve (Panjkovich & Svergun (2017) Bioinformatics 4(11):1944-1946. doi: 10.1093/bioinformatics/btx846.)
To better account for the flexibility of Trx-MYOT, the ensemble optimization method (EOM) was employed. For this a large pool of 10 000 independent models with random conformations was generated by connecting the available high resolution structures of individual domains with randomized linkers represented by chains of dummy residues. The pdb codes used are 2KDG.pdb for myotilin Ig1, 2KKQ.pdb for myotillin Ig2 and 1T7P.pdb for the Trx tag. With a genetic algorithm-based selection, representative models were picked resulting in the best fit to the data with χ2 = 1.0. Overall, the EOM-selected models tend to be more compact than those in the random pool, whereby the most extended models are not selected. The volume fractions of the selected and deposited models (starting from top to bottom) were 10%, 10%, 40%, 20%, 10% and 10% respectively.
|
|
 s, nm-1
s, nm-1
 Rg, nm
Rg, nm