|
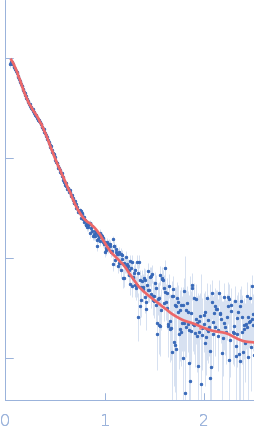
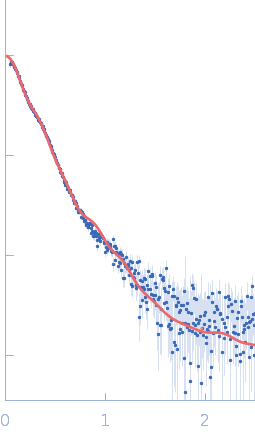
Synchrotron SAXS data from solutions of Human MUC2 mucin C-terminal dimer in 25 mM HEPES, 100 mM NaCl, 10 mM CaCl2, pH 7.4 were collected on the BM29 beam line at the ESRF (Grenoble, France) using a Pilatus3 2M detector at a sample-detector distance of 2.8 m and at a wavelength of λ = 0.0826 nm (I(s) vs s, where s = 4πsinθ/λ, and 2θ is the scattering angle). In-line size-exclusion chromatography (SEC) SAS was employed. The sample (100 μl at 1.8 mg/ml) was injected and ran into an Agilent Bio SEC-3 300Å column at a flow rate of 0.3 ml/min at 25°C. Scattering data was collected throughout the entire SEC column elution with exposure time of 1sec/frame (for 540 frames total). Data reduction and 1D scattering intensities of the sample were performed as per standard BM29 beam line protocols. Twenty-eight data frames selected from the sample elution peak were selected and processed (averaged and buffer subtracted) using CHROMIXS and primusqt from the ATSAS 3.0.0 package. The radius of gyration (Rg), forward scattering (I(0)), maximum particle dimension (Dmax) and the distance distribution function were determined using primusqt and GNOM. The calculated molecular weight (MW) of this molecule including the most probable glycans is around 240 kDa for the dimer. SDS-PAGE suggests a MW <150 kDa monomer and >250 kDa dimer for this molecule. The MW estimation from the SAXS data (primusqt) varied depending on the method used: Qp = 444.7 kDa, MoW= 208.5 kDa, Vc= 267.8 kDa, Size & Shape=309.7 kDa. MW estimation of highly glycosylated proteins is still challenging. According to Guttman et al. 2013, the MoW approach is more accurate than the autoporod estimation for glycosylated proteins. However, Vc and Size & Shape methods also suggest reasonable values for the dimer. The models displayed in this entry included: Top. Single ab initio model obtained by dummy atom modeling using DAMMIN. Middle/Bottom: Combined model of cryoEM data and alphafold, modloop and CHARMM-GUI predictions. The model was compared with the SAXS data using FoXS and CRYSOL, respectively.
Notes: The model used for this comparison was produced by the combination of the CryoEM structure (7QCL), an alphafold prediction from the terminal part, and a combination of ModLoop and CHARMM-GUI predictions of missing loops and the glycans previously predicted by mass spectroscopy (van der Post et al. 2014 Proteome Res.). The model is missing 30 amino acids at the N-terminus.
|
|
 s, nm-1
s, nm-1